의연방09) PSA 분석1(매칭)
Propensity Score Analysis I (matching)



📜 제목으로 보기✏마지막 댓글로
- 학습목표
- Motivation(하는 이유)
- PS 추정 방법
- PS Matching
- R 실습
- 정리하기
- R 고급 실습(ps matching with datacleaning-dplyr, MatchIt, tableone-CreateTableOne, geepack-geeglm, broom-회귀계수를 OR로 변환) 포함됨
- 위험요소-질병간의 상관관계 -> 인과관계를 살펴보고 있는데
- 상관관계가 곧 인과관계를 의미하지는 않는다
- 상관관계가 관측될 경우, 아래를 확인해야한다.
- 이 상관관계가 진짜인가?
- 우연이나 잘못된 연구디자인에 의한 것인가?
- 상관관계가 진짜로 판단되는 경우
- 인과관계가 성립하는가?
confounding에 의해 생겨난 상관관계인가?
- 연구 디자인 단계에서 confounding을 최대한 제거한다
- 랜덤화 -> RCT를 하는 것
- 매칭
- 데이터 분석 단계에서 confounding 효과를 보정한다
- Stratification
- confounder 값별로 그룹을 다시 나눠서 분석
- Adjustment
- multivariable model로 분석하는 것
- Propensity score analysis
- confounding 효과를 보정하기 위해서
- (마치 RCT에서 나온 것 같이) 데이터를 가공하여, X값을 랜덤하게 배정했을 때 관측될 법한 virtual data를 만들어 분석한다
- Stratification
- Confounding이 존재하는 관찰연구의 데이터를 ->
RCT의 데이터처럼 confounding이 없도록 가공하여 분석하는 것 -
Propensity score (PS): (치료군/대조군 비교 연구에서) 치료군에 속할 확률- 위험요소 - 질병 or 치료방법 - 질병 간의 관계를 살펴볼 때
-
위험요소O/X or 치료O/X2군으로 나누고,질병 발생 빈도를 살펴보게 된다.
-
-
2군으로 나누었을 때, 그 중에 1군(치료군)에 속할 확률을 PS라 한다.
- RCT에서는, 모든 환자에 대해서 PS = 0.5(치료군 0.5 -> 대조군도 0.5)
- 관찰연구에서는 PS(치료군에 속할 확률)는 알려지지 않은 (일정한 확률이 없는)미지의 값
- 관찰연구에서 PS는 환자 특성(or 치료자, 환자가 속한 지역/병원)에 따라 달라짐
- 위험요소 - 질병 or 치료방법 - 질병 간의 관계를 살펴볼 때
- PS를 사용하는 이유?
- PS에 condition을 하고나면,
baseline characteristics(환자 특성)와treatment selection(치료군이냐 대조군이냐)이 독립이 된다. - 무슨 얘기냐?
- PS가 비슷한 환자들끼리 모아놓으면, 치료군/대조군의 baseline characteristics 분포가 비슷해진다
- 원래는 환자 특성(baseline characteristics) -> 환자가 치료군에 속할 확률(treatment selection)에 영향을 주게 되는데
- 환자 특성을 일일히 확인해서 비슷한 사람들끼리 모아놓는게 아니라, 환자 특성에 영향 받는 PS만 알고 있다면, PS를 기준으로 비슷한 환자를 모아두기만 하면, 환자 특성 분포가 비슷해진다
- PS에 condition을 하고나면,
- consistency(컨시스턴시)
- 환자가 받은 치료법에 따라서, 치료군이든 대조군이든 관측될만한 outcome(질병)이 있다. 그것이 real world에서 관측한 것과 outcome(질병)이 같다
- 환자의 potential outcome = 실제로 관측된 outcome
- no unmeasured confounder
- 모든 confounder를 알고 있고 관측했다는 가정
- Positivity : 치료군이 될 확률>0, 대조군이 될 확률>0
- 어떤 환자든지 한쪽에만 속하는게 아니라 2군 각각 속할 확률이 존재한다
- no misspecification of PS model
- PS score 추정모델이 잘못되어선 안된다.
- 3번 제외 다른 가정들은 만족시키기 어렵다
- 모든 통계모델은 많은 가정아래 존재한다.
- 그런데 항상 만족시키는 것은 아니다. 현실을 완벽하게 반영은 X
- PSA에 대한 사람들의 의문점
- “Confounder는 공변량으로 Multivariable regression model에 넣으면 보정 되는 것 아닌가요?”
- 이것도 유효한 방법이나, treatment effect가 조금 다르게 나온다.
- “Confounder는 공변량으로 Multivariable regression model에 넣으면 보정 되는 것 아닌가요?”
- Regression에서 추정하는 효과 :
conditional treatment effect-
각 환자 개개인에서의 평균 효과
-
- PSA와 RCT에서 추정하는 효과 :
marginal treatment effect-
모집단 전체에서의 모든 환자의 평균 효과
-
- 두 개는 뭐가 다른 것일까?
-
Conditional treatment effect in Regression
- “
한 환자에게 혈압강하제를 투여하는 실험을 여러번 반복할 때, 평균적으로 수축기 혈압을 10만큼 낮춘다”- 여기서 -10이 Conditional treatment effect이다.
- 이 환자에게 약을 투약하는 실험을 여러번 반복해서 평균적으로 수축기혈압을 10만큼 낮춘다.
- 개인에서의 평균
- “
-
Marginal treatment effect in PSA, RCT
- “
고혈압 환자 전체에서 평균적으로 수축기 혈압을 10만큼 낮춘다”- 여기서 -10이 ConditMarginal treatment effect이다.
- 각 환자에게 낮춰진 수축기혈압을 모두 구해, 모든 고혈압 환자에서 평균적으로 10만큼 낮춘다.
- 전체에서의 평균
- “
-
한 환자당 10만큼 낮추면(conditional) -> 평균적으로 전체에서도 10만큼 낮춰질 수 밖에 없다.(marginal)
- 이 것이 가능한 상황은,
- 현재
연속현 변수(수축기혈압)의 평균으로 추정하거나 - 아니면,
이분형 변수에서 치료군vs대조군 비율의 차이(Risk Differenece)로 계산할 경우에 - conditional == marginal treamment effect 로 항상 같다
- 현재
- 하지만,
- 효과가
연속변수의 평균 차이 또는 이분형 변수의 비율차이(RD)가 아니라OR 또는 생존분석의 Hazard Ratio로 측정되는 경우도 있디.- conditional tx effect ≠ marginal tx effect로 다르다.
- 이 것이 가능한 상황은,
-
그럼 무엇을 써야할지 어떻게 결정할까?
-
내가 추정할 치료효과가
RCT에서 나타나는 결과와 유사한가?- yes라면, Multivariable regression가 아닌 Propensity score analysis가 더 적합하다 판단하고 Marginal treatment effect in PSA, RCT 를 구하면 된다.
-
내가 추정할 치료효과가
- PS를 어떻게 추정할 것인가
- PS가 비슷한 환자들은 confounder가 없는 상태라고 하는데, 비슷한 환자들은 어떻게 모아서 치료효과를 추정할 것인가?
PS model의 설명변수
- propensity score를 구하기 위해 regression model에 어떤 설명변수를 넣어야할까?
- PS 개념상으로는
이 사람이 치료군에 속할지/대조군에 속할지 결정(X, 치료여부)에 영향을 끼치는 변수들을 모두 설명변수에 변수들(아래 그림 중 가운데에서 C)을 넣는 것이 맞는 것 같지만... - X(어느군에 속할지 == 치료여부) -> Y(outcome)
- PS 개념상으로는
- 추가로, 여러 시뮬레이션을 통한 여러 연구결과:
-
치료군/대조군 결정(X)에 영향을 끼치지 않더라도 outcome(Y)에
영향을 끼치는 변수들(3번째 그림)은 넣는 것이 좋다라고 연구 결과가 나왔다.
- 분산을 감소시킴 = p-value가 낮아짐 && 신뢰구간 좁아짐 등의 효과**
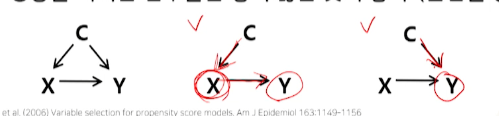
- X(치료여부)에는 영향을 끼치지 않더라도 Y(outcome)에 영향을 끼친다면 넣자.
- 분산을 감소시킴 = p-value가 낮아짐 && 신뢰구간 좁아짐 등의 효과**
-
outcome(Y)에 영향을 끼치지 않으면서 치료군/대조군 결정(X)에
영향을 끼치는 변수들은 넣지 않는 것이 좋다 (분산을 증가시킴)
- PS정의상으로는 넣으면 좋을 것 같지만
- 연구결과 넣지 않는 것이 더 좋다고 나옴
- 가운데 그림의 C -> 넣지 않는 것이 좋다라고 연구결과가 나온 것도 있다.
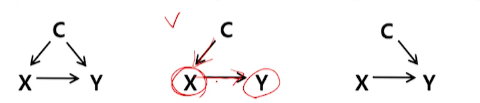
-
치료군/대조군 결정(X)에 영향을 끼치지 않더라도 outcome(Y)에
영향을 끼치는 변수들(3번째 그림)은 넣는 것이 좋다라고 연구 결과가 나왔다.
- 그럼 어떤 것을 PS의 regression model에 어떤 설명변수를 넣어야할까?
-
보통 Table 1에 들어가는 baseline characteristics는 치료군/대조군 결정(X)과 outcome(Y) 둘 다에 영향을 준다
- → 다 포함하는 것이 좋다
table1의 기본특성은 거의다 confounder로 생각된다. 다 포함시키면 된다.
- PS model은 overfitting 문제가 없다
- 원래 다변수 모델에서는
- 설명변수가 많아지면 -> 데이터에 비해 모델이 복잡해져 overfitting이 발생하지만
-
PS model에서는 그런 overfitting 문제가 없다
- → 설명변수 개수에 제한이 없다
-
PS model은 우리가 알고 싶어하는 것에 대한 최종 model이 아니라 confounder 보정의 중간단계이기 때문에, 넣고 싶은만큼 많이 넣어도 된다.
- 조금이라도 confounder 같다고 의심되는 변수는 모조리 포함시키면 된다.
- 원래 다변수 모델에서는
-
절대 포함하면 안되는 변수
치료군/대조군 결정 이후 관측된 변수- PS의 정의= 치료군에 속할 확률
- 치료군/대조군 결정되기 이전의 변수들로 판단되어야한다
- 군 결정이후 발생된 변수는, 과거(군 결정)에 영향을 주면 안된다.
- cf) 관측만 결정 이후에 되었을뿐이지, 그 변수값 자체는 치료군vs대조군 결정이전에 결정된 경우도 있다.
- 예) 2가지 수술방법에 대한 비교 연구
- 수술이후 조직을 biopsy한 결과 -> 치료군vs대조군 결정이후 = 수술방법 결정이후 관측되었지만
- 수술이전에 이미 내부장기상태는 결정된 상태라고도 볼 수 있다.
- 예) 2가지 수술방법에 대한 비교 연구
- PS model을 만들어서 PS를 추정했다고 가정하자
- 그 이후 다음 단계 분석은 어떻게 해야할까?
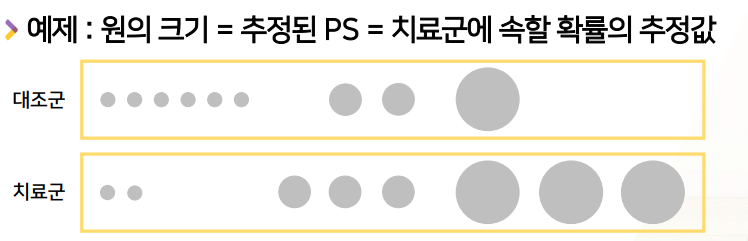
- PS가 비슷한 환자별로 치료군:대조군 1:1 쌍을 지어주는 것
- 짝이 지어진 환자들만 분석하고, 짝이 안지어지면 분석에 사용안한다
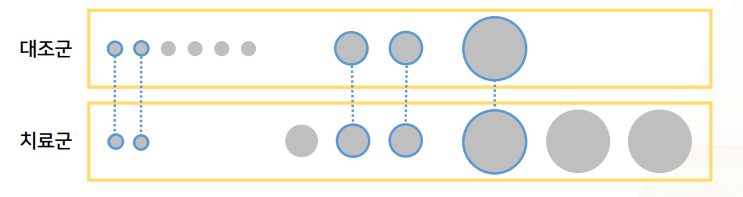
- 2군에 걸쳐 짝이 지어졌다 = 분포가 동일해진다. = 전체적으로 PS가 비슷해짐 = 다른 기본특성도 비슷해짐 = confounding이 제거된다.
- 장점 : 직관적으로 이해가 쉽다
- 단점 : 짝지어지지 않는 환자의 데이터를 잃어버린다
- n은 클수록 좋은데, 모아진 데이터를 버려야하니 큰 단점
- 짝이 지어진 환자들만 분석하고, 짝이 안지어지면 분석에 사용안한다
-
With replacement vs. without replacement
- With replacement: 매칭해서 짝 지은 다음 -> 복원 추출처럼 다른 환자랑 매칭가능하게 하는 것
- 통계적으로 더 좋은 성질을 가지지만, 여러번 들어갈 수 있어서 -> 사람들이 껄끄럽게 생각함
-
withoutreplacement: 비복원추출 -> 매칭후 분석풀에서 재고- 실제로 많이 사용되는 것
- With replacement: 매칭해서 짝 지은 다음 -> 복원 추출처럼 다른 환자랑 매칭가능하게 하는 것
-
Greedy vs. optimal matching vs. full matching
-
Greedy: 한명씩 순차적으로 매칭- 가장 많이 쓰임.
- optimal: 전체적 2군의 PS가 비슷해지도록 한번에 매칭
- full: 버림없이 모든 환자를 매칭
-
-
1:1 vs. n:1
- 대조군이 너무 많을 경우 -> 대조군:치료군 = 4:1까지 매칭된다.
-
오스틴이 밝힌 가장 좋은 매칭 방법은?
-
nearest neighbor matching(=Greedy) withcaliper- 치료군 환자에 대해 가장 PS가 비슷한 환자를 뽑더라도, 가장 비슷한 PS를 뽑더라도, 치료군의 PS와 차이가 많이 나는 대조군이 뽑을 수 있다.
-
PS차이의 한계를 정해놓은 것 =
caliper-
logit of PS을 먼저 계산한다log 𝑃𝑆/(1-𝑃𝑆)
- logit of PS의 표준편차를 계산하고, 여기에 0.2를 곱한 값을 caliper로 생각한다.
- 0.2 *
SDoflogit of PS
- 0.2 *
- 위의 caliper를 치료군-대조군 matching시 PS차이 한계점으로 생각(넘으면 버림)하고 nearest neighbor matching을 돌리면, confounder로 인한 치료효과 추정치에 생기는 bias가 99%가 제거된다.
-
-
-
이로 인해
nearest neighbor(=Greedy) matchingwithcaliper를 가장 많이 쓴다.
- 매칭을 한 뒤, 잘 매칭되었는지 확인하기 위해
- PS matching -> 목적:
confounding이 없어졌는지==baseline characteristic 차이가 정말 줄어들었는지를 확인해야한다 - 즉, baseline characteristics의 차이가 줄어들었는지 평가해야한다
- PS matching -> 목적:
- 이 때,
매칭된 dataset에 대해- 잘못된 방식으로서
- 치료군과 대조군의 각 baseline characteristics의 요약통계량 계산 후
-
2군의 비교이므로p-value로 계산하면 될까?- 2군 비교시 p-value가 익숙해져 있어서 매칭데이터도 p-value로 비교하는 경우가 많다 -> 좋은 방법이 아니다.
- 잘못된 방식으로서
-
매칭된 dataset을 p-value로 비교하면 왜 잘못된 방식일까?- p-value는 sample size에 영향을 받지만
- matching의 경우, 대부분 매칭되지 않는 환자들이 제외되어
- original sample size보다 줄어들게 되고
- power가 줄어든다 -> 두 군간의 차이가 있었더라도 -> p-value는 높게 나올 수 있다.
- p-value가 높게 나온 것이, 줄어든 sample size 때문인데, 마치 matching이 잘되어서 p-value가 나온 것처럼 잘못된 impression을 줄 수 있다.
- p-value로 비교한다는 것은
- 2군의 sample에 대해 2개의 큰 모집단이 있다는 것을 가정한 것이다.
- 2개 모집단을 비교할 수 없어서 random sample을 뽑아, 2개 모집단을 추론하는 것이 p-value이다.
- 그러나 우리는 matching 했다
- 2군 뒤의 2개 모집단에 대한 관심은 잃은 상태다.
- 2개 모집단은 애초에 baseline characteristic이 다르기 때문에, confounding으로 인한 치료효과 분석이 어렵다고 생각해서 인위적으로 matching을 하는 것
- 인위적으로 baseline characteristic를 맞춘, 인위적은 dataset을 보는 것
- 더이상 sample로 모집단을 추론(p-value)하는 것이 아니라
sample 자체에 관심을 가지게 되는 것이므로p-value로 2군 비교는 적합하지 않다. - 개념적으로 아예 맞지 않는 것
- 그렇다면 옳은 매칭후 평가 방법은?
- matching후 평가방법은 여러가지가 있다. 그 중에서 사용하기 편하고 이해하기 쉬운 방법인
Standardized mean difference를 통해 평가해보자
- matching후 평가방법은 여러가지가 있다. 그 중에서 사용하기 편하고 이해하기 쉬운 방법인
Standardized mean difference (SMD)
-
연속형 변수의 SMD
- 복잡해보이지만,
치료군의 평균(X_bar_tx)와대조군의 평균(X_bar_tx)의 차이에서 -
치료군과 대조군의 표준편차를 잘 합친 것(
pooled SD)으로 나누어,데이터의 변동을 고려했을 때의 평균 차이
- 복잡해보이지만,
- 이분형 변수의 SMD
- 2 군의 비율 차이를 적당한 표준편차 값으로 나눈 것으로 생각하자
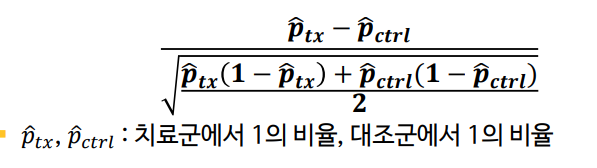
- 2 군의 비율 차이를 적당한 표준편차 값으로 나눈 것으로 생각하자
- 두 군간 평균의 차이가 pooled SD의 몇배인가
- SMD < 0.1~0.2 이면 작은 차이로 간주
- 매칭 이후 매칭변수들(PS model에 사용된 설명변수들)에 대해 SMD를 계산하고
- 대부분 0.1미만이면 좋다
-
몇개는 0.1을 넘어
모두 0.2 미만이면 balance가 잘 맞는 것으로 평가 - 이정도면,매칭된 dataset에서 치료군과 대조군 사이에 baseline characteristic차이가 무시할만한 차이이다
-
분석을 하려면, 우리가 애초에 궁금한
치료효과에 대해 평가해야한다.-
치료여부-질병여부와의 association이 얼마나 강한가?를 평가하는measure of association를 이용해야한다. - 여러가지 방법이 있었다.
-
OR말고 RD(risk diffrence) = attributable risk라던가 relative risk를 계산할 수 있지만 - OR이 가장 계산이 쉽고, 편리하기 때문에 대표로 사용해보자.
-
-
-
매칭된 치료군과 대조군에서 결과변수의 차이
- 이분형 결과변수의 경우:
OR과OR의 95% 신뢰구간,OR이 1인지 아닌지에 대한 p-value정도를 사용한다
- 이분형 결과변수의 경우:
-
보통의 방법을 사용하면 되는가?
- 매칭된 dataset은, 매칭되면서 짝끼리 서로 비슷하게 맞춰놓은 상태기 때문에, 환자가 서로 독립이라고 할 순 없다. 그래서 대부분의 통계방법은 서로 독립이라고 가정을 하는데, 일반적인 통계방법으로 분석하면 안된다. -> 매칭된 짝끼리는 독립이 아니라는 점을 고려해서 분석하는 것이 좋다는 것이 다수 의견이다.
-
매칭된 짝은 서로 독립이 아니라는 점을 고려하여 분석한다
- 이분형 결과변수의 경우:
-
McNemar’s test: p-value를 계산해주지만, OR자체를 계산해주지 않는다.- OR 계산하려면 logistic regression을 해야한다.
-
conditional logistic regression- case-control study에서 사용했던 것. 매칭된 짝끼리 비슷한 점 = 독립이 아니라는 점을 이용할 수 있다.
-
logistic regression with correlation structure (GEE)- 일반 logistic regression을 하되, 매칭된 짝끼리의 correlation을 허용하면서 모델을 fit하는 것
- GEE model이라고 하는데, 사실은 계산방법이다.
- GEE 계산을 통한 로지스틱 회귀분석을 통해 OR을 계산하고, 신뢰구간을 구하면 된다.
-
- 이분형 결과변수의 경우:
-
소수의견 : RCT에서 얻어진 데이터처럼 만들었기 때문에, RCT처럼 독립성을 가정하고 분석해야 한다는 관점도 존재한다
- conditional logistic regression이나 logistic regression with correlation structure (GEE)을 수행할 때
- 결과 변수는
질병여부 - 설명 변수는
치료군vs대조군 여부가 되는데 -
애초에 confounder라고 생각했던 공변량들은 설명변수로 포함할 필요가 없게 된다.
- 왜냐면,
PS matching을 통해 공변량 효과를 이미 제거된 상태가 되었기 때문이다. - 즉, multivarable이 아니라 univariable의 로지스틱 회분석해서 OR(오즈 레이시오)를 계산하면 된다는 것이다.
- 왜냐면,
- 결과 변수는
- 그럼에도 불구하고, 공변량으로 포함시켜 계산하는 방법도 존재하는데
doubly robust하다라는 이름을 얻게 되었다.- PS model이 잘못됬거나 아니면, 최종 모델로 생각한 회귀분석이 잘못되었어도, OR 추정치가 unbiased하게 되는 좋은 성질을 갖게 된다고 한다.
- 하지만, 매칭 분석은 univariable model만 쓰면 된다.
-
MatchIt패키지를 이용한 PS matching- matchit() 함수 이용
- Austin이 추천한
nearest neighbor matching with caliper= 0.2*SD of logit of PS 을 구현하는 방법 실습
-
tableone패키지를 이용한 SMD 계산- CreateTableOne() 함수 이용
- 매칭후 balance 계산을 위해 SMD를 계산.
-
geepack패키지를 이용한 OR 추정- geeglm() 함수 이용
- 매칭된 데이터셋에서 GEE방법으로 OR를 계산해주는 패키지
- PS matching이외에 여러가지 매칭이 가능한 패키지이나 필요한 것만 설정하기
matchit(formula, data, method, replace, ratio, distance, link, caliper, std.caliper, m.order)
- formula : 매칭하려는 그룹 (치료군/대조군)~ 매칭변수1+매칭변수2+...+매칭변수d
-
치료군이냐 대조군이냐 그룹변수~confounder라고 의심해서 PS model에 넣고 싶은 변수들을 더하기로 연결
-
- data : 데이터셋 이름
- method : 매칭방법. “nearest”라고 입력하면 nearest neighbor matching을 수행한다
- replace : 재복원여부.
- 디폴트는 FALSE (비복원표집, 한번 매칭된 환자는 다시 매칭되지 않는다)
- without replacement = 한번 매칭된 환자는 다시 매칭하지 않는 것이 default
- Ratio : 매칭비율 (치료군 1명당 매칭되는 대조군 환자 수). 디폴트는 1.
- 치료군 1명당 매칭될 대조군의 수
- distance=“glm”
- matching 중에 Propensity Score matching을 이용하는 옵션
- link=“logit”
- caliper의 기준을 정하는데, PS의 logit를 거리측도로 사용하여 매칭을 수행한다
- caliper=0.2
- caliper 기준에 곱해줄 값을 입력
- std.caliper = TRUE
- caliper 의 표준편차를 기준으로 매칭
- 위 4가지를 다 설정해야, austin의 PS login의 SD의 0.2를 caliper로 사용한 매칭을 실시하게 된다
- m.order=“random”: 매칭을 수행하는 순서
-
입력하지 않으면 거리측도 값 = PS값이 큰 개체부터 순서대로 매칭한다.
- 그러나 PS score 큰 수대로 매칭해봐야 장점이 없다. 랜덤으로 매칭하도록 선호된다.
- “random”으로 입력하면 랜덤한 순서로 매칭하는데, 이 경우 matchit() 함수 실행 직전에
set.seed()함수로 seed number를 정해주어야 같은 결과를 재생산할 수 있다. (그렇지 않으면 matchit() 함수 실행 때마다 다른 매칭결과가 나온다
-
입력하지 않으면 거리측도 값 = PS값이 큰 개체부터 순서대로 매칭한다.
CreateTableOne(vars, data, strata)- 임상과학 논문의
Table 1에 해당하는 표를 편리하게 만들 수 있는 함수- vars : 표에 들어갈 변수 이름의 벡터
- data : 데이터셋 이름
- strata : 비교할 그룹을 나타내는 변수
- 스트레이라를 입력해주면, 그룹을 나눠 그룹별 요약통계량과 그룹별 p-value가 나온다.
- CreateTableOne()으로 만든 객체를 print() 안에 넣으면 표가
인쇄된다
- smd : TRUE로 입력하면 SMD를 계산해준다
- test : FALSE로 입력하면 p-value를 인쇄하지 않는다
geeglm() from geepack
geeglm(formula, id, family, corstr, std.err, data)- GLM(generalized linear model)을 GEE(generalized estimating equation) 방법으로 correlation structure를 주면서 fit하는 함수
- formula : 결과변수~설명변수
- fitting하고 싶은 model 입력
-
id : cluster를 나타내는 변수
- PS maching의 경우 같이 매칭된 자료의 경우
matching id를 넣어주면 된다. - 내가 생각하기에 correlate되어있는 클러스트를 나타내는 변수를 넣는다.
- PS maching의 경우 같이 매칭된 자료의 경우
-
family : GLM의 종류. 로지스틱 회귀분석의 경우 “binomial”
- 로지스틱 회귀를 설정 옵션
- corstr : cluster 내에서의 correlation structure.
- 어떤 correlation structure를 줄 것이냐
-
“exchangeable”: cluster내에서 모든 개체 쌍의 correlation이 동일할 경우
- 매칭된 환자(치료군 1명에 대조군 1~4)는 같은 cluster내에 있으며, 매칭 환자들 중에 아무 2명씩 짝을 지어도 항상 correlation같다는 옵션
- 가장 무난한 옵션
- “independence”: cluster내에서 모든 개체가 서로 독립일 경우
- 매칭된 cluster환자들 내(치료군+매칭된 대조군(들))이 서로 독립이라고 가정하고 싶을 때 쓰는 옵션이나
- 매칭되었으니까 independent할텐데, 왜 쓸까?
- 어떤 복잡한 통계적 방법에 의해 이 방법이 더 타당하다고 주장/연구결과가 있어서 옵션으로 언급만
- std.err : standard error 계산방식.
- 디폴트 “san.se”는
robust estimator방식으로 계산하게 되는데, 일반적인 방식이 아니라 데이터가 매칭=독립이 아니다라고 가정하여, 더 보수적으로 계산한 estimator다 - 95% 신뢰구간과 p-value 계산시 영향을 끼친다
- robust estimator를 써야 더 보수적인 결과가 나오며 default옵션이다.
- 디폴트 “san.se”는
- data : 데이터셋 이름
# install.packages("MatchIt") -> 설치 안되서 터미널에서 아래와 같이
# conda install -c conda-forge r-matchit
# - ocnda로 검색해서 안되서, stackoverflow에서 얻은 설치주소
# - https://stackoverflow.com/questions/71041174/installing-r-package-matchit-in-anaconda-environment
library(MatchIt)
# - matchIt 다른 곳에서 설치 -> R을 4로 업데이트 -> 추가적인 것 수동 업데이트
library(dplyr)
library(tableone)
data("lalonde")
head(lalonde)
# treat age educ race married nodegree re74 re75 re78
# 치료여부 / / 교육 / 결혼여부 / nodgree 고등학교졸업/ 년도별 연소득
# 의료데이터는 아니나,, 가정하고 한다.
dat1 <- lalonde %>%
# outcome 변수이름을 간단히 y로 둔다.
# re78이 5000달러를 넘으면 [질병여부 발생여부]로 임시로 가상으로 만든다.
mutate(y=ifelse(re78 > 5000, 1, 0))
head(dat1)
dat1 <- lalonde %>%
mutate(y=ifelse(re78 > 5000, 1, 0))
# - y도 0 or 1로 바꿔줬는데, 연속형변수(숫자)처럼 되어있을 것이다.
# - 연속형 변수는 min/max가 나와있다.
# - 치료여부(treat) 와 질병여부(y)가 모두 연속형 변수로 되어있다.
# 그외
summary(dat1)
dat1 <- lalonde %>%
mutate(y=ifelse(re78 > 5000, 1, 0)) %>%
# vars(변수들), list(~하고싶은작업(.)) . 자리에 변수들이 들어간다.
mutate_at(vars(treat, married, nodegree, y),
list(~as.factor(.)))
summary(dat1)
# install.packages("haven")
# install.packages("forcats")
# install.packages("hms")
tobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = dat1)
tobj
# -> treat 변수별로 보도록 strata를 treat로 지정해준다.
tobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = dat1, strata = "treat")
tobj
tobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = dat1, strata = "treat")
print(tobj)
# treat가 0 , 즉 대조군에 대한 4가지 confounder변수의 요약통계량이 나온다.
# 치료군에 대해서도 나온다.
# 치료군 185, 대조군 429명의 정보도 나온다.
# p-value를 보고, 2군간의 confounder( baseline characteristic)의 차이를 표1을 만들어서 본다.
# -> "p가 낮으면 두 군간의 유의미한 차이"가 있다는 뜻이다.
# ->age married nodegree 는 2군간에 유의미하게 차이가 난다.
tobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = dat1, strata = "treat")
print(tobj, smd = T)
# 0.2를 넘는 것(married)은 두 군간에 상당히 다르다는 것을 의미한다.
mobj <- matchit(treat ~ age+educ+married+nodegree , # formula : 치료여부 ~ confoiunder들
data = dat1, # dataset
method = "nearest", # 매칭 방법: austin의 nearest ~
distance = "glm", # 매칭 중 PS matching
link = "logit",
replace = F, # 매칭된 환자 비복원
capliper = 0.2, std.caliper = T, # caliper 지정
m.order = "random", #매칭순서 -> default방법 안좋음. random으로 지정
)
# 뭐가 들었는지 일단 summary 안에 넣어본다.
summary(mobj)
# 썩 보기 좋은게 아님.
# 맨 마지막만 보고, 몇 쌍이 매칭 되어있는지 확인한다.
# Sample Sizes:
# Control Treated
# All 429 185
# Matched 185 185
# Unmatched 244 0
# Discarded 0 0
# -> 185쌍이 매칭되었다.
m.data <- match.data(mobj)
summary(m.data)
# ps를 의미하는 distance가 보인다.
# subclass -> 짝 지어진 번호. 1인 환자 -> 다른데 또 1인 매칭환자가 있다
# - 즉, matching Id를 의미한다.
mobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = m.data, strata = "treat")
print(mobj, smd = T)
# print( , test = F)
print(mobj, smd = T, test = F)
# 매칭후에는 smd가 0.2보다 다 작다
mobj <- matchit(treat ~ age+educ+married+nodegree , # formula : 치료여부 ~ confoiunder들
data = dat1, # dataset
method = "nearest", # 매칭 방법: austin의 nearest ~
distance = "glm", # 매칭 중 PS matching
link = "logit",
replace = F, # 매칭된 환자 비복원
capliper = 0.2, std.caliper = T, # caliper 지정
m.order= "random",#매칭순서 -> default방법 안좋음. random으로 지정
)
m.data <- match.data(mobj)
tobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = m.data, strata = "treat")
print(tobj, smd = T, test=F)
# 원래는 seed를 정해주지 않으면, 매칭 n수가 바귄다.
# -> 여기서는 m.order = random을 바꾸면 SMD가 바껴서 적용은 되는 것 같다.
# -> 혹은 seed가 자동적용되었을 가능성이 있다.
set.seed(2022)
mobj <- matchit(treat ~ age+educ+married+nodegree , # formula : 치료여부 ~ confoiunder들
data = dat1, # dataset
method = "nearest", # 매칭 방법: austin의 nearest ~
distance = "glm", # 매칭 중 PS matching
link = "logit",
replace = F, # 매칭된 환자 비복원
capliper = 0.2, std.caliper = T, # caliper 지정
m.order= "random",#매칭순서 -> default방법 안좋음. random으로 지정
)
m.data <- match.data(mobj)
tobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = m.data, strata = "treat")
print(tobj, smd = T, test=F)
# SMD가 다 0.2 이하라서 2군간의 confounder차이가 사라지도록 매칭된 데이터가 완성되었다.
set.seed(2022)
mobj <- matchit(treat ~ age+educ+married+nodegree , # formula : 치료여부 ~ confoiunder들
data = dat1, # dataset
method = "nearest", # 매칭 방법: austin의 nearest ~
distance = "glm", # 매칭 중 PS matching
link = "logit",
replace = F, # 매칭된 환자 비복원
capliper = 0.2, std.caliper = T, # caliper 지정
m.order= "random",#매칭순서 -> default방법 안좋음. random으로 지정
ratio=2, # 치료군(1) 한명당 대조군 2명을 붙인다.
)
m.data <- match.data(mobj)
tobj <- CreateTableOne(vars=c("age", "educ", "married", "nodegree"),
data = m.data, strata = "treat")
print(tobj, smd = T, test=F)
# 만약, 185명의 2배가 아니라 더 적게 매칭되었다면?
# -> caliper가 있어서, PS가 비슷한 환자를 찾지 못해서 1명만 매칭된 경우가 포함되어있다.
library(geepack)
library(broom)
# -> 결과변수를 다시 연속형 변수로 매핑해줘야한다.
head(m.data$y == 1)
head(as.double(m.data$y == 1))
obj <- geeglm(as.double(y == 1) ~ treat,
data = m.data, #
id = subclass, # subclass칼럼에 정의된 각 매칭id를 제시하여 correlation이 있다는 것을 알려준다.
family = "binomial", # 로지스틱 회귀분석 명시
corstr = "exchangeable", # correlation structrue 암기...
std.err = "san.se", # default라 안써도 되는 에러 계산시, robust estimator 사용
)
# 회귀분석 결과는 기본적으로 summary에 넣으면 된다.
summary(obj)
# Coefficients:
# Estimate Std.err Wald Pr(>|W|)
# (Intercept) -0.01093 0.10426 0.011 0.917
# treat1 -0.17337 0.18076 0.920 0.337
# treat1에 있는 Estimate -> 회귀계수 -0.17337 과 p-value 0.337
# -> 강의랑 달라서 아래 스샷을 바탕으로...
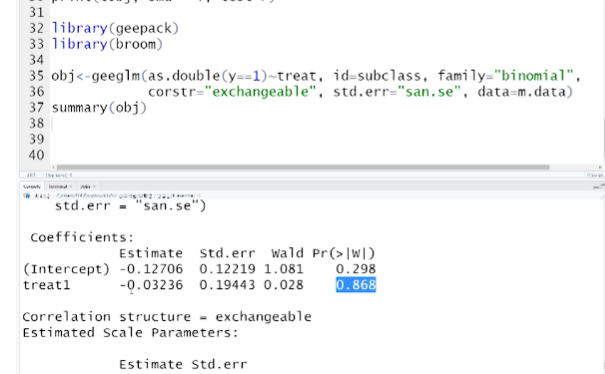
# treat1의 p-value가 0.05보다 크다 == treat1이라는 변수는 회귀변수 결과변수에 별로 안중요하다?!
# -> treat여부가 질병발생(outcome발생) 비율에 유의미한 영향을 주지 않는다?!
# 주의) 여기서의 p-value는 회귀계수 자체의 p-value이지, OR이 아니다.
# -> 우리가 해석할 것은 treat의 회귀변수의 p-value가 아니라 OR를 구해야한다.
# broom패키지의 tidy()함수로 회귀분석결과 -> OR를 바로 구할 수 있다.
tidy(obj, exponentiate = T, conf.int = T)
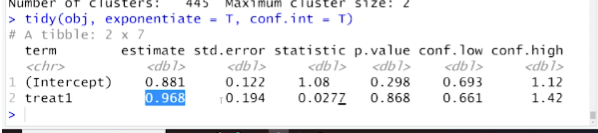
# conf.low ~ conf.high는 OR의 95%신뢰구간 하한~상한이다.
- Propensity score analysis (PSA)는 confounding이 존재하는 관찰연구의 데이터를
RCT의 데이터처럼 confounding이 없도록 가공하여 분석하는 방법이다. -
Propensity score (PS)는치료군에 속할 확률을 의미한다. - PS는
보통 로지스틱 회귀분석으로 추정하며, PS 모델에는outcome에 영향을 끼칠 것으로 생각되는 변수를 모두 넣는 것이 좋다. 단, 치료군/대조군 결정이후에 관측된 변수는 넣으면 안된다. - PS 추정 이후 분석 방법에는 matching, stratification, weighting, covariate adjustment 등이 있다.
- PS matching 후 치료군과 대조군의 balance는 standardized mean difference (SMD)로 평가할 수 있다.
- PS matching된 데이터는 matching을 고려하여 분석한다.
01 다음 중 Propensity score analysis에 대한 설명으로 틀린 것은?
-
관찰연구 자료의 confounding을 없애는 방법 중 하나이다.
-
Propensity score를 추정하는 모델은 설명변수 하나당 샘플사이즈 15가 필요하다.
-
최종결과변수(환자의 outcome)에 영향을 끼치는 변수들은 PS 추정 모델에 넣는 것이 좋다.
-
치료군/대조군이 결정된 이후에 관측된 변수들은 PS 추정 모델에 넣지 않는다.
- 정답 : 2
- 해설 : PS 추정 모델은 overfitting의 문제에 구애받지 않고 많은 수의 설명변수를 포함할 수 있다.